Neu bei immundefekt.de: Fragen (FAQ) und Antworten
Was ist ein Immundefekt?
Unser Immunsystem dient der Abwehr von Infektionserregern wie Bakterien, Mykobakterien, Viren, Pilzen und Parasiten. Zudem trägt es dazu bei, dass Autoimmunerkrankungen, Autoinflammation, Lymphoproliferation, Allergien und maligne Erkrankungen verhindert werden. Bei einem Immundefekt können eine oder mehrere dieser Funktionen gestört sein. Dann spricht man von einem Immundefekt.
Was für Immundefekte gibt es?
Immundefekte können angeboren (primärer Immundefekt = PID, engl. inborn errors of immunity) oder erworben (sekundärer Immundefekt = SID) sein. Angeborene Immundefekte lassen sich in der Mehrzahl auf Gendefekte zurückführen. Derzeit sind mehr als 500 verschiedene Gendefekte bekannt. 485 davon wurden zuletzt 2022 offiziell klassifiziert .
Sekundäre Immundefekte können z.B. durch Tumorerkrankungen wie chronisch-lymphatische Leukämie (CLL) oder multiples Myelom entstehen, aber auch durch die Therapie mit immunsuppressiven oder zytostatischen Medikamenten. Zudem können auch andere schwere Allgemeinerkrankungen oder schwere Ernährungsstörungen die Funktionen des Immunsystems beeinträchtigen.
Häufigkeit
Wie viele Menschen haben einen angeborenen Immundefekt (Immunschwäche, Abwehrschwäche, primäre Immundefekte = PID)?
Diese Frage lässt sich wissenschaftlich nicht eindeutig beantworten. Deutsche Register liefern vermutlich zu niedrige Zahlen, weil zum einen PID oft nicht als solche diagnostiziert werden, zum anderen bekannte PID-Patienten nicht an die Register gemeldet werden. Eine Studie aus den USA hat daher elektronische Gesundheitsdaten verwendet, um der Frage näher zu kommen. Danach leiden in den USA 6/10000 Personen an einem PID. Die Studie hat sicher methodische Schwächen. Man darf sie aber als Hinweis nehmen, dass auch in Deutschland die Anzahl der diagnostizierten PID-Patienten weit unter der Anzahl der tatsächlich vorhandenen Patienten liegt.
Entstehung
Wie entsteht ein angeborener Immundefekt (PID)?
Derzeit sind gut 500 verschiedene Gendefekte bekannt, die zu einem Immundefekt führen. Pro Monat kommt ca. 1 neuer Gendefekt hinzu. Mit dem Suchbegriff „Neuer PID“ (exakter Ausdruck!) können diese mit der Suchmaschine bei immundefekt.de aufgelistet werden. Daneben gibt es eine große Zahl von Patienten mit einem PID, wo die Pathogenese noch nicht bekannt ist. An diesen Patienten wird weltweit intensiv geforscht.
Klinik
Wie macht sich ein angeborener Immundefekt (PID) bemerkbar?
Im Zentrum der klinischen Probleme der meisten Patienten steht die Anfälligkeit gegenüber Infektionserregern. Daneben können aber auch andere Manifestationen auftreten, entweder allein oder in Verbindung mit krankhafter Infektionsanfälligkeit. Die folgende Abb.
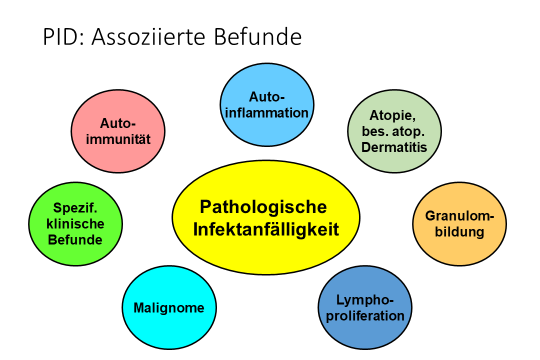
und das Video geben eine Übersicht.
Diagnostik
Welcher Arzt untersucht das Immunsystem?
Im Prinzip kann jeder Arzt einfache Untersuchungen des Immunsystems veranlassen, meist wird es aber der Haus- oder Kinderarzt sein.
Wie stellt der Arzt einen Immundefekt fest? Welche Blutwerte sind wichtig? Welche nicht?
Der behandelnde Arzt wird zunächst prüfen, ob es Warnzeichen für einen Immundefekt gibt. Ist das der Fall, wird er eine Basisdiagnostik zur Orientierung über einfache Funktionen des Immunsystems durchführen, die man hier findet. Diese Blutwerte sind wichtig. Dagegen sind Messungen von diversen Vitaminen und Spurenelementen (Zink, Selen…) für die Basisdiagnostik verzichtbar.
Wo gibt es Zentren in Deutschland, die sich mit Immundefekten besonders gut auskennen?
Über die Webseite des deutschen Ärztenetzwerks FIND-ID kann man über diese Adresse Immundefektzentren für Kinder und Erwachsene finden, auch über die Webseite der Arbeitsgemeinschaft Pädiatrische Immunologie (API) bei.
Therapie
Welche Therapiemöglichkeiten gibt es?
Wenn eine genaue, möglichst genetische Diagnose gestellt ist, hat der Arzt verschiedene Behandlungsmöglichkeiten.
- Substitution fehlender Plasmaproteine (z.B. Immunglobuline, C1 Inhibitor)
- Antimikrobielle Substanzen (Antibiotika, Antimykotika, antivirale Substanzen) zur Akut-Therapie oder Langzeitprophylaxe
- Gezielte immunpharmakologische Therapien (z.B. Hemmung überaktivierter Enzyme oder Signaltransduktionselemente bei GOF Mutationen)
- Zytokine, hämatopoetische Wachstumsfaktoren (z.B. G-CSF, Interferon-γ)
- Immunsuppressive Maßnahmen (z.B. bei Patienten mit Autoimmunerkrankungen)
- Stammzelltransplantation (z.B. bei SCID, bestimmten CID, CGD u.a.)
- Somatische Gentherapie (zugelassen bisher nur für SCID bei ADA-Mangel)
- Kombinationen verschiedener Maßnahmen
Ist ein angeborener Immundefekt heilbar?
Nur nach Stammzelltransplantation oder somatischer Gentherapie kann es zur Korrektur eines Immundefekts i.S. einer Heilung kommen. Alle anderen Verfahren gelten als Therapie, die nur so lange wirkt, wie sie auch verabreicht wird.
Sind Maßnahmen zur "Stärkung des Immunsystems" wirksam?
Bei Patienten mit PID sollten nur erwiesenermaßen wirksame Therapieverfahren eingesetzt werden. Homöopathie, Naturheilverfahren, Nahrungsergänzungsmittel, Probiotika, Multivitaminpräparate und andere in der Werbung lautstark angepriesenen Substanzen sind oft teuer und nutzlos. Dagegen ist eine ausgewogene und gesunde Ernährung sinnvoll. Nur bei einzelnen Patienten mit spezifischen Problemen müssen bestimmte Vitamine (z.B. B12) zur Therapie eingesetzt werden.
Impfungen
Können PID-Patienten geimpft werden?
Bei den meisten PID können Impfungen ohne Probleme durchgeführt werden, auch wenn z.T. mit eingeschränktem Schutz gerechnet werden muss. Warnen muss man bei bestimmten PID vor Lebendimpfungen (Masern, Mumps, Röteln, Rotaviren, Gelbfieber, BCG u.a.), weil es dabei zu schwerwiegenden Impfkomplikationen kommen kann. Je nach Immundefekt müssen Impfungen daher individuell geplant werden. Weitere Informationen zu diesem Thema finden sich auch in einer Veröffentlichung der STIKO .